Graph Neural Networks finding drugs for cancer target HER2
Find the source code here: https://github.com/littlemountainman/GraphCNN_CancerHER2
Abstract
The HER2 receptor tyrosine kinase plays a crucial role in various cancers. Identifying potent inhibitors of HER2 is essential for therapeutic intervention. Virtual screening (VS) offers a method to expedite drug discovery, but traditional methods struggle to capture the complexity of protein-ligand interactions. This study investigates the potential of Graph Neural Networks (GNNs) for predicting binding affinities of compounds with the HER2 kinase. Leveraging GNNs’ message-passing mechanism to model the intricate spatial relationships between HER2 and potential drug candidates. This approach aims to streamline the VS process for HER2, facilitating the identification of promising drug leads and potentially enabling the repurposing of existing drugs. Finally ,presenting 5 compound which should be further investigated in-vitro.
Introduction
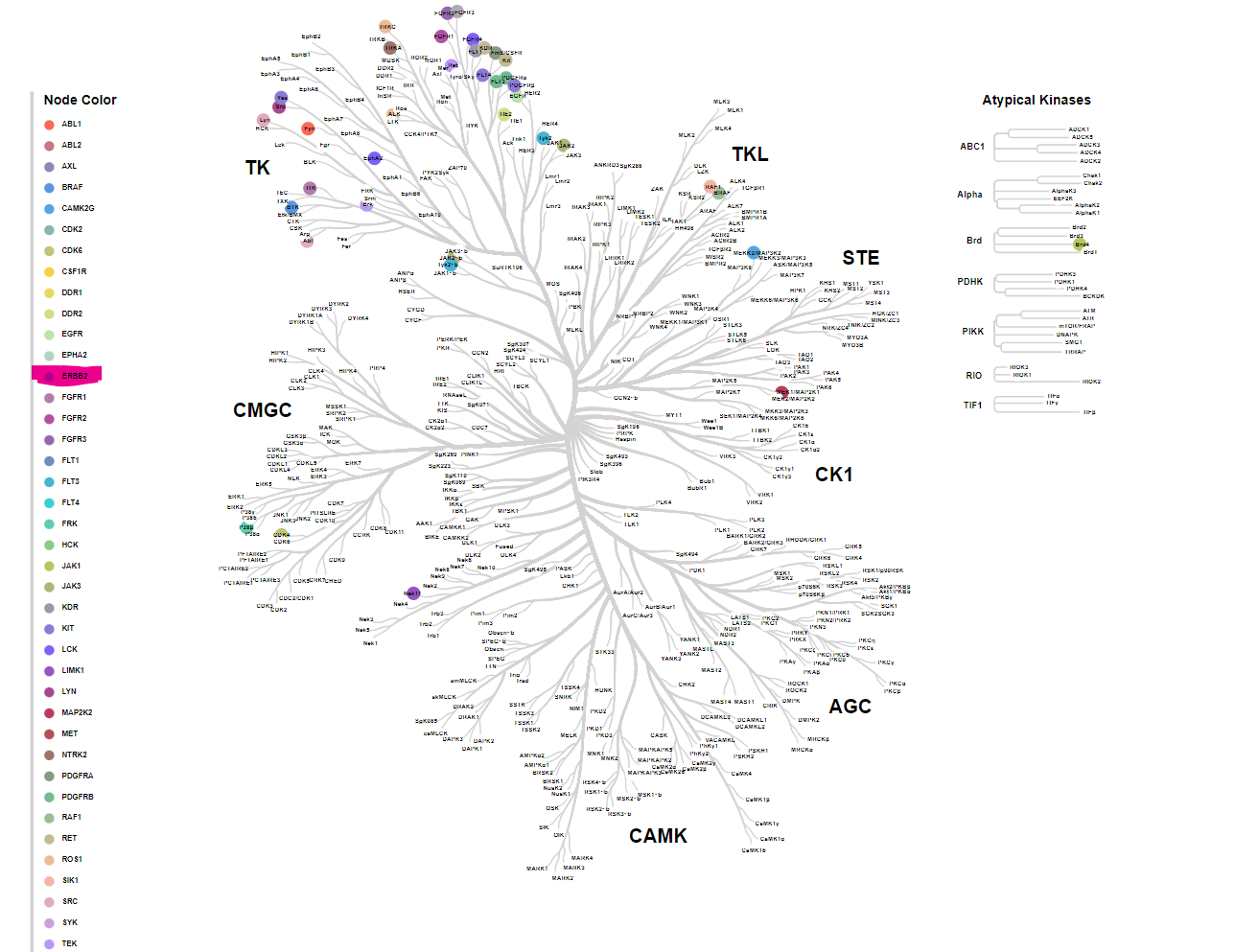 31.07.2024 Taken from here
31.07.2024 Taken from here
{kind=link}
The human kinome, encompassing receptor tyrosine kinases (RTKs) such as the ERbB family, play a critical role in various cellular processes, including growth and programmed cell death (apoptosis). Aberrations in these pathways, particularly mutations leading to the overexpression of specific RTKs, contribute to cancer development. Among these, the HER2 receptor, encoded by the ERBB2 gene, is a well-established oncogenic target owing to its role in uncontrolled cell proliferation and resistance to apoptosis. Consequently, there is significant interest in identifying potent inhibitors of HER2 for therapeutic intervention.
Virtual screening (VS) offers a powerful approach for expediting drug discovery by computationally identifying pre-existing bioactive compounds with promising binding affinities to target proteins. Traditionally, VS methods rely on docking simulations or machine learning models that utilize simplified representations of molecules. However, these methods often struggle to capture the complex 3D interactions and spatial relationships that are crucial for binding affinity prediction.
In recent years, Graph Neural Networks (GNNs) have emerged as a transformative deep learning technique for analyzing graph-structured data . GNNs excel at capturing complex interdependencies within graphs, making them ideally suited for modeling protein-ligand interactions. This study investigates the potential of GNNs for predicting binding affinities of compounds with the HER2 kinase.
By leveraging the message-passing mechanism of GNNs, we aimed to develop a model that effectively captures the intricate spatial relationships between the HER2 protein structure and potential ligand molecules. This approach has the potential to streamline the virtual screening process for HER2, facilitating the identification of promising drug candidates for further development, potentially leading to the repurposing of existing drugs.
Dataset
As the foundation of our training pileline served KiBA. Across 52,498 chemical compounds and 467 human kinase targets, resulting in a total of 246,088 scores, which aim to integrate $K_i$, $K_d$ and $IC_{50}$ by optimizing consistency among the metrics. No further modifcations to the scores neither the target sequence, nor the compound’s SMILE string have been conducted.
Data preperation
This section describes our process for transforming the linear notation for molecules, known as SMILES strings, into a graph representation that may be used for a variety of machine learning applications. The molecule’s atom-level characteristics and structure are both encoded in this graph form.
We define a function, denoted by $f(a)$, that takes an atom object a as input and generates a feature vector $x_a$capturing its properties. This vector incorporates the following information: One-hot encoding of the atom’s element type (Carbon, Nitrogen, Oxygen, etc.), One-hot encoding of the atom’s degree (number of bonded atoms),One-hot encoding of the atom’s total implicit hydrogens, One-hot encoding of the atom’s implicit valence (e.g., 0-10),Binary flag indicating if the atom is aromatic.To address potential unseen element types during graph construction, we introduce the function $f_{oh}(x,\mathcal{S})$, which extends the standard one-hot encoding $f_{oh}(x,\mathcal{S})$ by assigning unknown input values (not present in the allowed set $\mathcal{S}$) to the last element in the set. This ensures consistent feature vector size regardless of the encountered atom type. Following the extraction of atom features, the next step involves constructing the graph representation of the molecule. This process entails two key stages, edge identification and graph construction. Edge identification extracts information about the chemical bonds connecting atoms within the molecule. These bonds represent the physical connections between atoms and are crucial for capturing the molecule’s structure in the graph. The identified connections are then stored in a set denoted by $\mathrm{E}$, representing the set of edges in the graph. Based on the identified edges $\mathrm{E}$, a graph $G=(\mathrm{V},\mathrm{E})$ is constructed. Here,$\mathrm{V}$ represents the set of nodes, corresponding to the atoms in the molecule.Finally, a list of edge indices I is generated. This list specifies the connections (edges) between nodes in the final graph.This process results in a graph representation G. Each node $v_i \in V$ in the graph corresponds to an atom $a_i$ within the molecule and possesses a feature vector $x_i$ encoding its properties.
Graph Neural Network
The proposed model for virtual screening utilizes a graph convolutional network (GCN) architecture to integrate previously processed graphs of ligand molecules. The model splits into two branches, a graph convolutional network branch to process the graph data and a protein sequence branch to process the protein sequences.
In the GCN branch, a GAT convolutional layer followed by a GCN layer is employed to capture node-node relationships and propagate information across the graph. Subsequently, global pooling is applied to extract graph-level features, which are then fed into dense layers.
For the protein sequence branch, an embedding layer first converts the protein sequence into a numerical representation. A bidirectional LSTM then captures sequential information in the protein sequence, and a 1D convolutional layer extracts features from the encoded sequence. These features are subsequently flattened and fed into dense layers.
Finally, the graph-level features and the protein sequence features are concatenated and fed into several dense layers with ReLU activation and dropout for learning a combined representation. The final output layer predicts the target-ligand binding affinity.
Results
Similarly to the processed training datasets KiBA, we processed the entire BindingDB database exactly the same way. Thus, resulting in searching a space of over 1.3 million possible compounds for our target. Listing our top 3 targets for HER2 inhibition below:
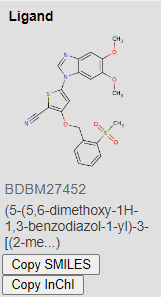
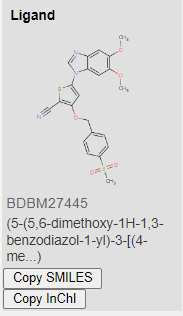
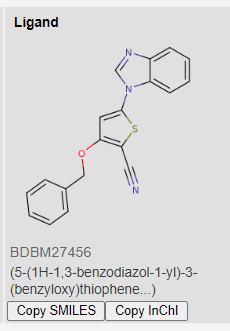
Images from BindingDB
Coincidentally, the 3 proposed compounds were previously developed by GSK for another kinase target. The resulting affinity values are 1.1945391, 2.8420124, 7.9635625, respectively for the 3 compounds above.
Low resulting values means that the drug is potent at low concentrations, and thus will show lower systemic toxicity when administered to the patient. Ergo, high binding affinity. Source
Update
Planning to release source-code soon!